Could LLMs help design our next medicines and materials?
The process of discovering molecules that have the properties needed to create new medicines and materials is cumbersome and expensive, consuming vast computational resources and months of human labor to narrow down the enormous space of potential candidates.
Large language models (LLMs) like ChatGPT could streamline this process, but enabling an LLM to understand and reason about the atoms and bonds that form a molecule, the same way it does with words that form sentences, has presented a scientific stumbling block.
Researchers from MIT and the MIT-IBM Watson AI Lab created a promising approach that augments an LLM with other machine-learning models known as graph-based models, which are specifically designed for generating and predicting molecular structures.
Their method employs a base LLM to interpret natural language queries specifying desired molecular properties. It automatically switches between the base LLM and graph-based AI modules to design the molecule, explain the rationale, and generate a step-by-step plan to synthesize it. It interleaves text, graph, and synthesis step generation, combining words, graphs, and reactions into a common vocabulary for the LLM to consume.
When compared to existing LLM-based approaches, this multimodal technique generated molecules that better matched user specifications and were more likely to have a valid synthesis plan, improving the success ratio from 5 percent to 35 percent.
It also outperformed LLMs that are more than 10 times its size and that design molecules and synthesis routes only with text-based representations, suggesting multimodality is key to the new system’s success.
“This could hopefully be an end-to-end solution where, from start to finish, we would automate the entire process of designing and making a molecule. If an LLM could just give you the answer in a few seconds, it would be a huge time-saver for pharmaceutical companies,” says Michael Sun, an MIT graduate student and co-author of a paper on this technique.
Sun’s co-authors include lead author Gang Liu, a graduate student at the University of Notre Dame; Wojciech Matusik, a professor of electrical engineering and computer science at MIT who leads the Computational Design and Fabrication Group within the Computer Science and Artificial Intelligence Laboratory (CSAIL); Meng Jiang, associate professor at the University of Notre Dame; and senior author Jie Chen, a senior research scientist and manager in the MIT-IBM Watson AI Lab. The research will be presented at the International Conference on Learning Representations.
Best of both worlds
Large language models aren’t built to understand the nuances of chemistry, which is one reason they struggle with inverse molecular design, a process of identifying molecular structures that have certain functions or properties.
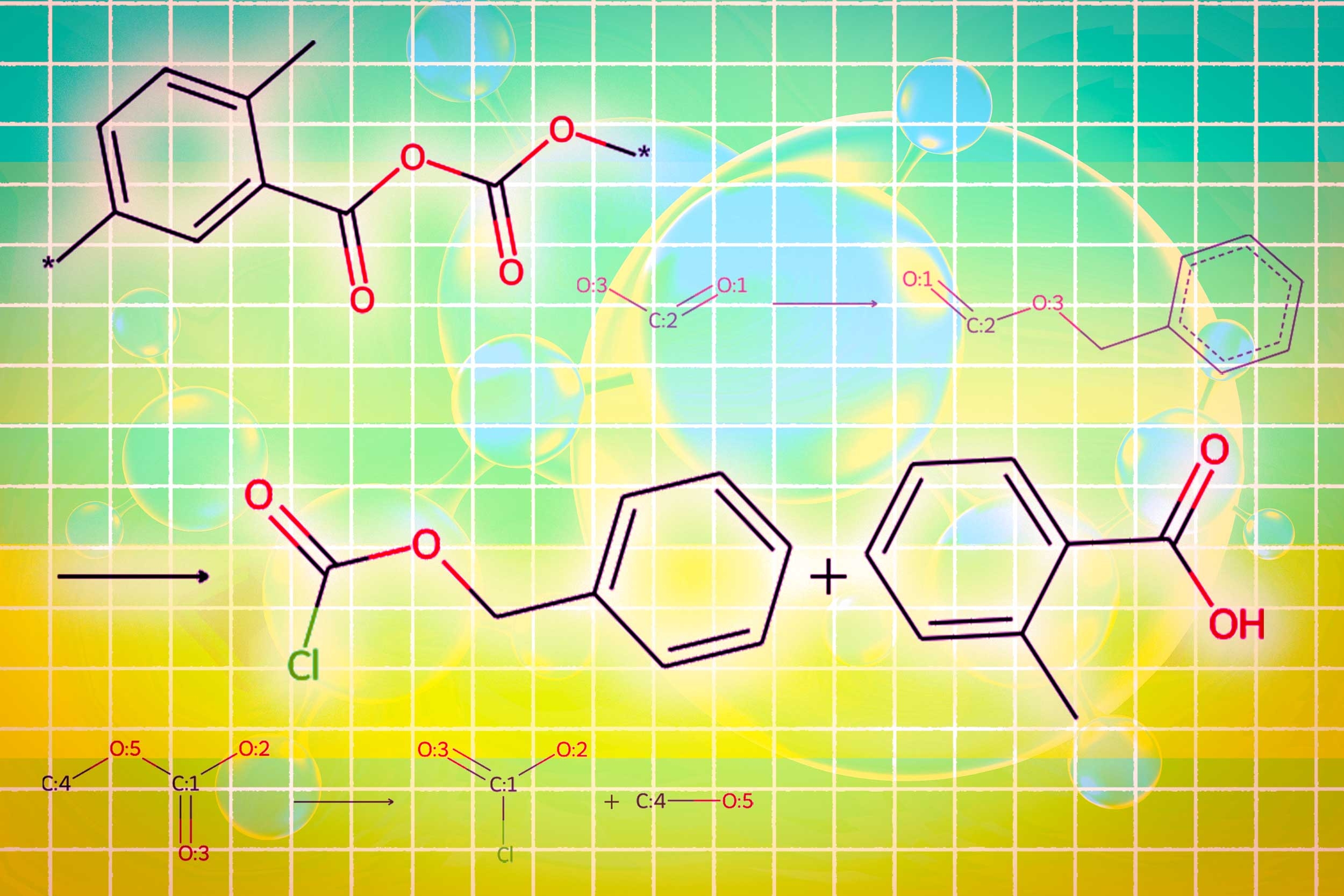
LLMs convert text into representations called tokens, which they use to sequentially predict the next word in a sentence. But molecules are “graph structures,” composed of atoms and bonds with no particular ordering, making them difficult to encode as sequential text.
On the other hand, powerful graph-based AI models represent atoms and molecular bonds as interconnected nodes and edges in a graph. While these models are popular for inverse molecular design, they require complex inputs, can’t understand natural language, and yield results that can be difficult to interpret.
The MIT researchers combined an LLM with graph-based AI models into a unified framework that gets the best of both worlds.
Llamole, which stands for large language model for molecular discovery, uses a base LLM as a gatekeeper to understand a user’s query — a plain-language request for a molecule with certain properties.
For instance, perhaps a user seeks a molecule that can penetrate the blood-brain barrier and inhibit HIV, given that it has a molecular weight of 209 and certain bond characteristics.
As the LLM predicts text in response to the query, it switches between graph modules.
One module uses a graph diffusion model to generate the molecular structure conditioned on input requirements. A second module uses a graph neural network to encode the generated molecular structure back into tokens for the LLMs to consume. The final graph module is a graph reaction predictor which takes as input an intermediate molecular structure and predicts a reaction step, searching for the exact set of steps to make the molecule from basic building blocks.
The researchers created a new type of trigger token that tells the LLM when to activate each module. When the LLM predicts a “design” trigger token, it switches to the module that sketches a molecular structure, and when it predicts a “retro” trigger token, it switches to the retrosynthetic planning module that predicts the next reaction step.
“The beauty of this is that everything the LLM generates before activating a particular module gets fed into that module itself. The module is learning to operate in a way that is consistent with what came before,” Sun says.
In the same manner, the output of each module is encoded and fed back into the generation process of the LLM, so it understands what each module did and will continue predicting tokens based on those data.
Better, simpler molecular structures
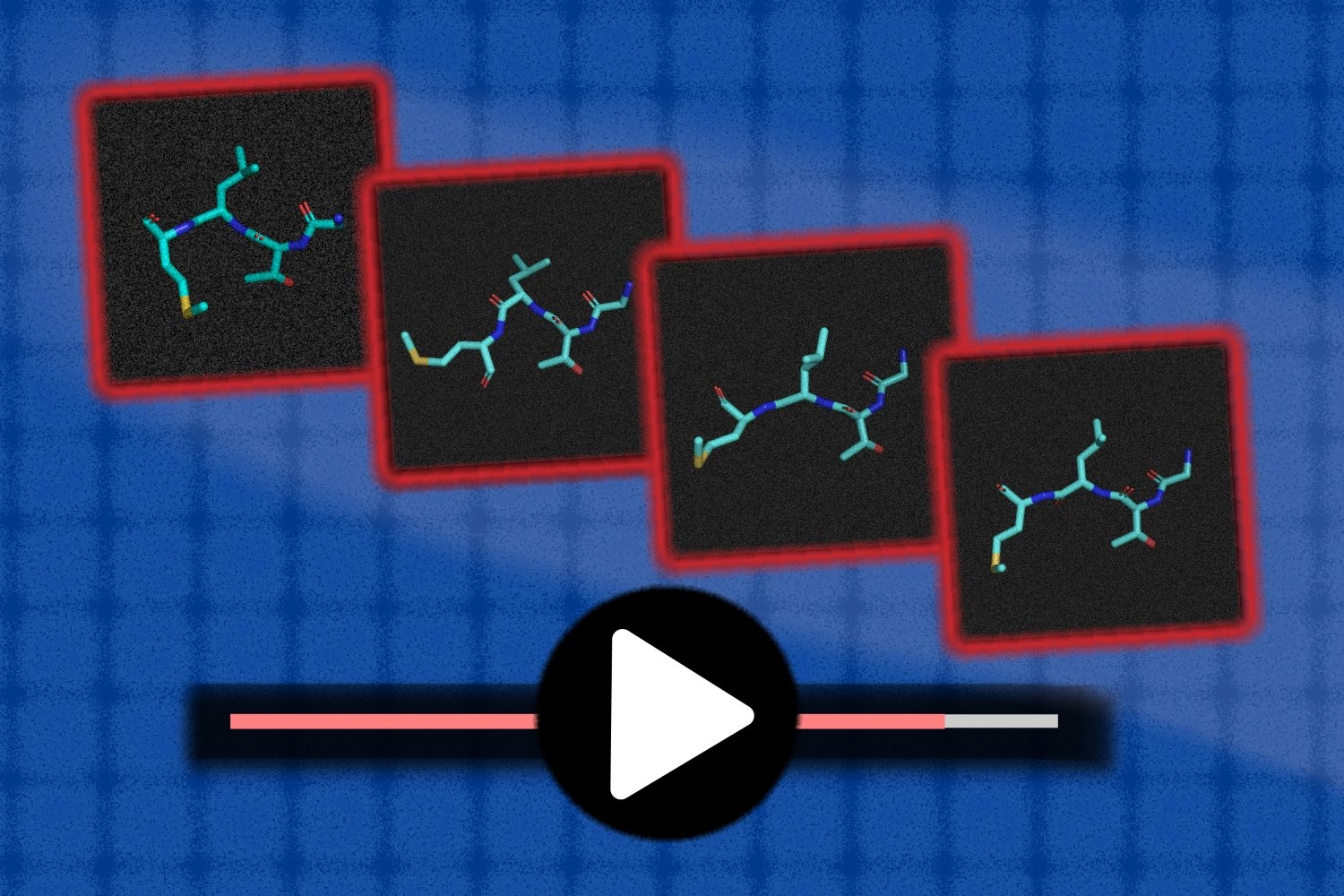
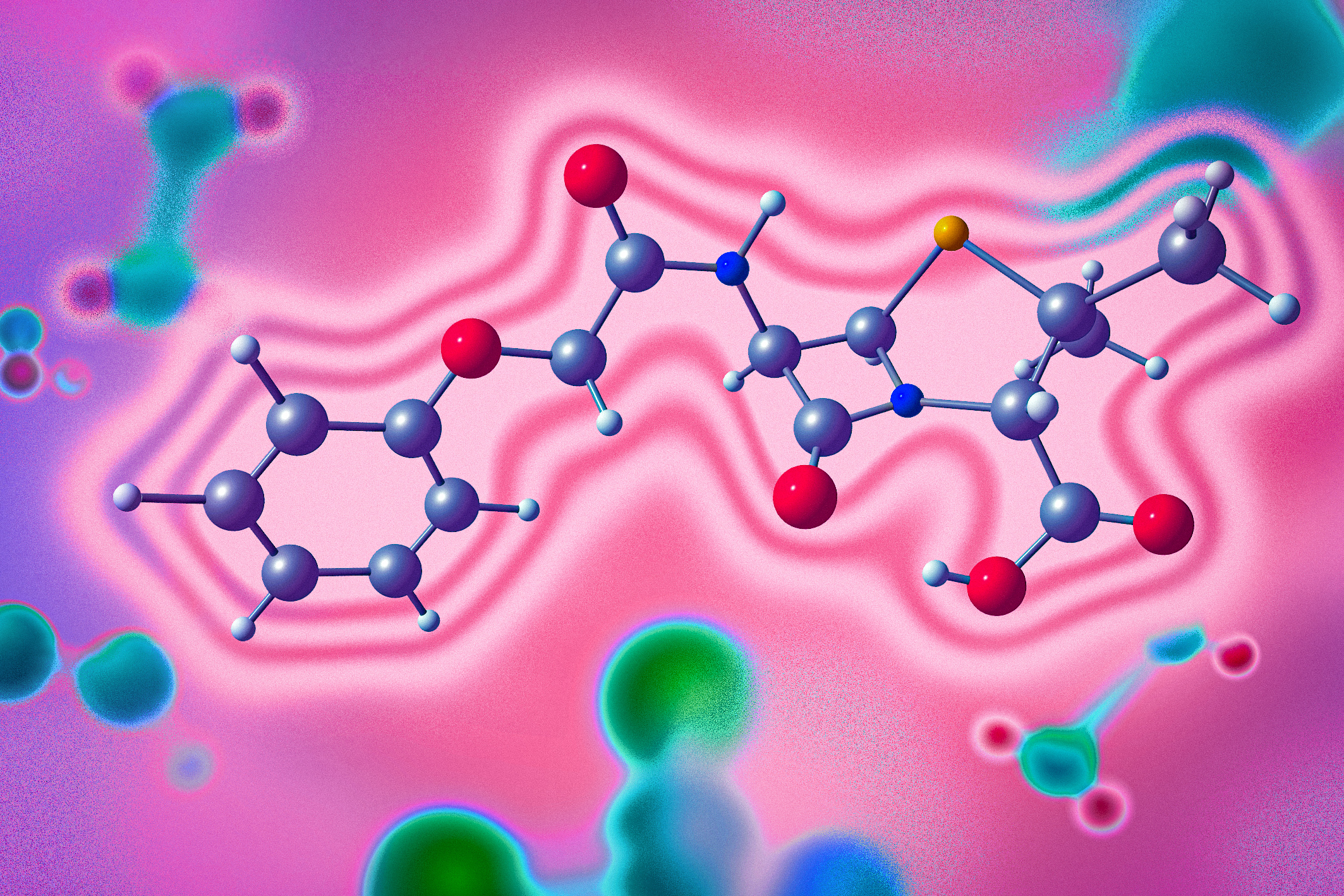
In the end, Llamole outputs an image of the molecular structure, a textual description of the molecule, and a step-by-step synthesis plan that provides the details of how to make it, down to individual chemical reactions.
In experiments involving designing molecules that matched user specifications, Llamole outperformed 10 standard LLMs, four fine-tuned LLMs, and a state-of-the-art domain-specific method. At the same time, it boosted the retrosynthetic planning success rate from 5 percent to 35 percent by generating molecules that are higher-quality, which means they had simpler structures and lower-cost building blocks.
“On their own, LLMs struggle to figure out how to synthesize molecules because it requires a lot of multistep planning. Our method can generate better molecular structures that are also easier to synthesize,” Liu says.
To train and evaluate Llamole, the researchers built two datasets from scratch since existing datasets of molecular structures didn’t contain enough details. They augmented hundreds of thousands of patented molecules with AI-generated natural language descriptions and customized description templates.
The dataset they built to fine-tune the LLM includes templates related to 10 molecular properties, so one limitation of Llamole is that it is trained to design molecules considering only those 10 numerical properties.
In future work, the researchers want to generalize Llamole so it can incorporate any molecular property. In addition, they plan to improve the graph modules to boost Llamole’s retrosynthesis success rate.
And in the long run, they hope to use this approach to go beyond molecules, creating multimodal LLMs that can handle other types of graph-based data, such as interconnected sensors in a power grid or transactions in a financial market.
“Llamole demonstrates the feasibility of using large language models as an interface to complex data beyond textual description, and we anticipate them to be a foundation that interacts with other AI algorithms to solve any graph problems,” says Chen.
This research is funded, in part, by the MIT-IBM Watson AI Lab, the National Science Foundation, and the Office of Naval Research.
© Image: MIT News; iStock